Michael S. Kay
Professor of Biochemistry
Protein Design, Viral Entry, Synthetic Biology, Chemical Protein Synthesis

Molecular Biology Program
Biological Chemistry Program
Education
B.A. Cornell University
M.D./Ph.D. Stanford University
Research
Our lab focuses on mirror-image peptides and proteins, which have great therapeutic potential because of their resistance to proteolysis. Our primary biological interest is developing D-peptide inhibitors against infectious diseases, particularly for the prevention and treatment of HIV and antibiotic-resistant bacterial infections, though we are now expanding into diverse therapeutic areas including cancer.
Mirror-image (D-peptide) Inhibitors
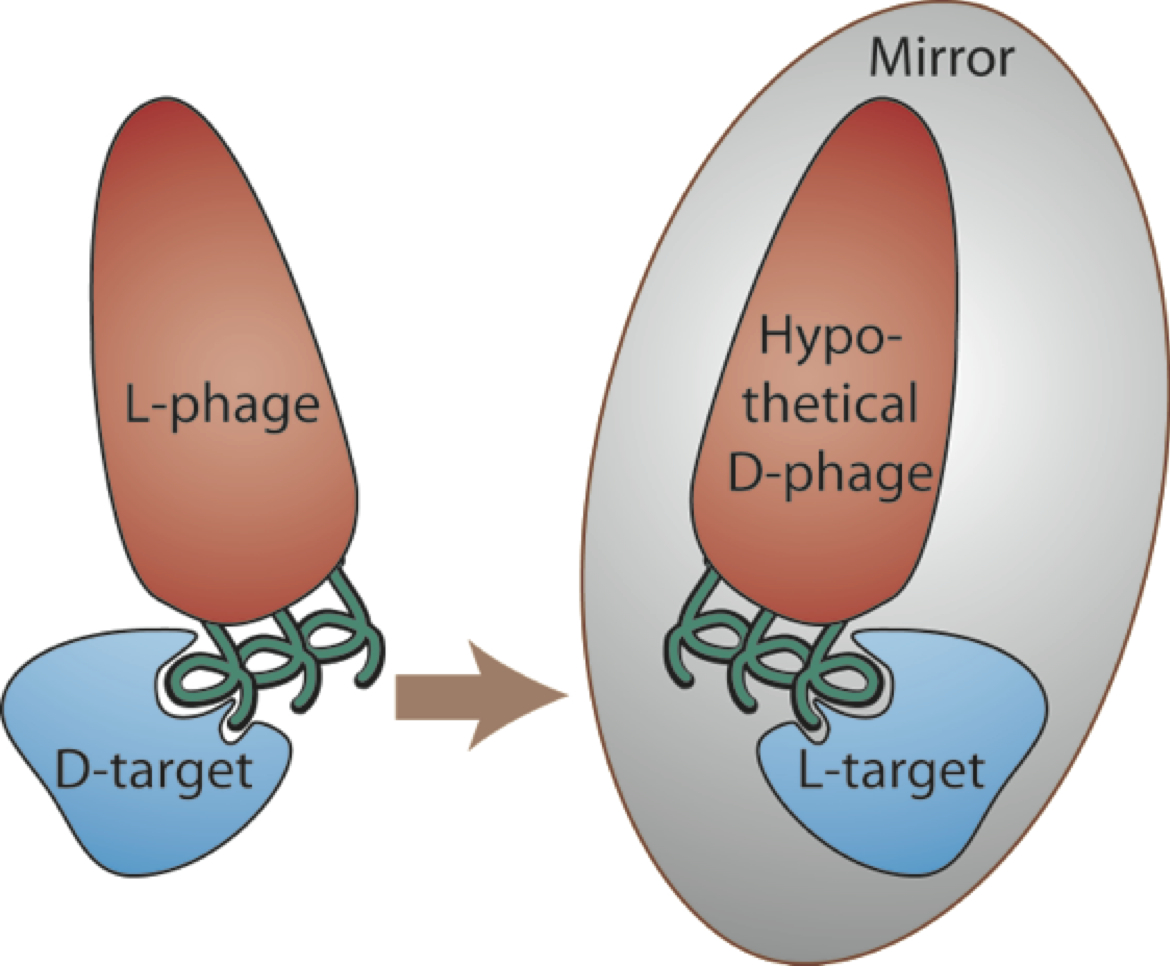
D-peptides are composed of mirror-image D-amino acids and have many potential advantages as therapeutics including low immunogenicity and protease resistance (allowing them to last much longer in the body than natural peptides). Since there are no natural examples to guide D-peptide design, we employ a high-throughput screening method called mirror-image phage display. In traditional phage display, billions of different peptide sequences are ‘displayed’ on the bacteriophage surface (each phage bearing a different peptide). High-affinity binders are identified through several rounds of screening against a specific target. In mirror-image phage display, the target of interest is synthesized from D-amino acids to produce a mirror-image target. L-peptides displayed on phage are then selected for binding to the D-target. By symmetry, D-versions of the discovered peptides will bind to the natural L-target (see figure).
D-peptide Inhibitors of HIV Entry
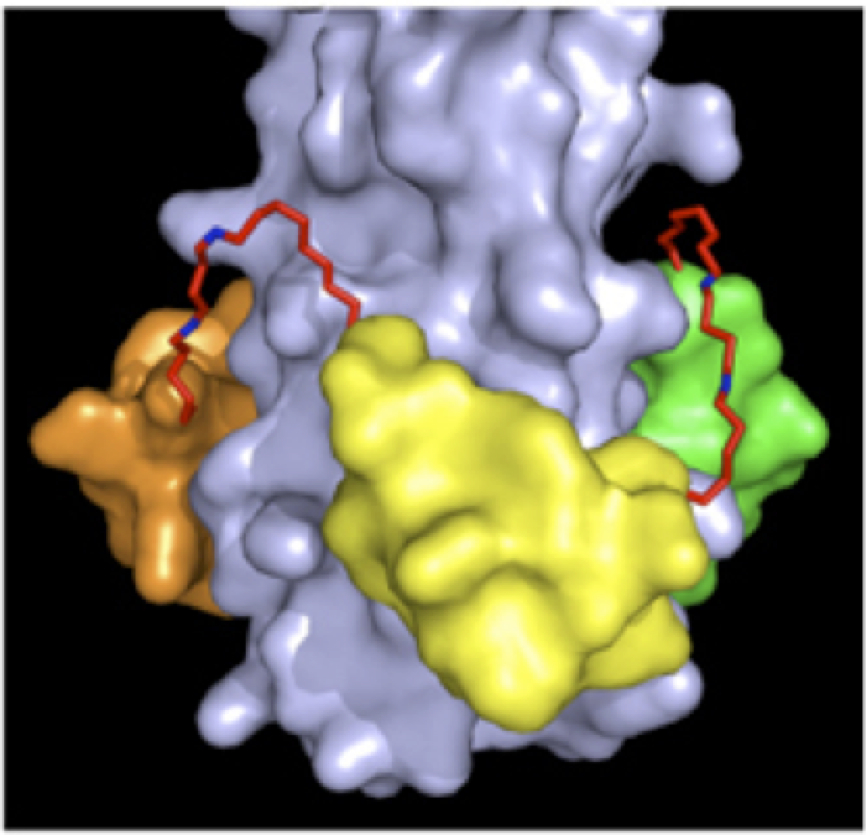
HIV membrane fusion (mediated by the HIV gp41 protein) has been identified as a promising target for inhibition. Fusion is initiated by CD4/coreceptor engagement, which triggers gp41 to extend and lance the target cell before collapsing into a six-helix “trimer of hairpins” that pulls the viral and target membranes together, leading to fusion. During this conformational transition, gp41 forms a transient pre-hairpin intermediate composed of a trimeric coiled coil. We are developing D-peptide inhibitors that bind to this intermediate and prevent HIV fusion and entry. Our inhibitor design also incorporates a unique "resistance capacitor" that provides a reserve of binding energy to combat potential resistance mutations (a major problem with HIV drugs).
Our most advanced D-peptide inhibitor of HIV entry, cholesterol-conjugated PIE12-trimer, has a long half-life, potently inhibits all major HIV strains, and has a high barrier to the emergence of viral resistance. This inhibitor is also highly effective as both a preventative and therapeutic agent in the gold-standard HIV animal model and has advanced to late-stage preclinical studies with clinical trials expected to start in 2020. See figure for a model of PIE12-trimer binding to the trimeric coiled coil.
Developing D-peptides as a Broad New Therapeutic Class
We are now applying our HIV experience to other therapeutic targets ideally suited for D-peptides. We are particularly interested in using D-peptides to treat challenging antibiotic-resistant bacterial infections in the gut and urinary tract. As a highly specific inhibitor, antibacterial D-peptides have the potential to block pathogenic bacteria without disrupting the healthy microbiome. As our ability to synthesize large D-protein targets continues to improve (see below), we are now able to tackle larger cellular protein targets with applications in inflammation and cancer.
The D. coli Project
Traditionally, D-peptide discovery efforts were limited to small target proteins that can be synthesized using solid-phase peptide synthesis (SPPS) (typically <100 residues). Our lab is developing tools to create larger chemically synthesized proteins and investigating mechanisms for folding these increasingly complex proteins. Recently, we reported the chemical synthesis of a record-length (312-residue) protein, DapA. Under physiologic conditions, DapA cannot fold on its own and requires the assistance of a bacterial chaperone (GroEL/ES). Using a synthetic version of D-DapA, we showed that GroEL/ES is ‘ambidextrous’, capable of folding mirror-image DapA. This result suggests that natural chaperones can be used to fold D-peptides for mirror-image drug discovery and synthetic biology applications.
Looking ahead, we are working to make D-protein production routine by developing new tools that facilitate chemical protein synthesis, such as solubilizing “helping hands” and an automated ligator (“Aligator”) program that predicts the most efficient synthetic routes for large proteins. Such tools are enabling us to pursue larger target proteins for mirror-image drug development. Our long-term goal is to synthesize a D-ribosome for easy production of any mirror-image protein as well as a key stepping stone towards ultimately creating a fully synthetic mirror-image organism that we have dubbed “D. coli”.
References
-
Fulcher JM, Petersen ME, Giesler RJ, Cruz ZS, Eckert DM, Francis JN, Kawamoto EM, Jacobsen MT, Kay MS. Chemical synthesis of Shiga toxin subunit B using a next-generation traceless "helping hand" solubilizing tag. (2019). Org Biomol Chem. 2019 Dec 3. doi: 10.1039/c9ob02012h.
-
Smith AR, Weinstock MT, Siglin AE, Whitby FG, Francis JN, Hill CP, Eckert DM, Root MJ, Kay MS. Characterization of resistance to a potent D-peptide HIV entry inhibitor (2019). Retrovirology. 16(1):28. doi: 10.1186/s12977-019-0489-7
-
Han H, Fulcher JM, Dandey VP, Iwasa JH, Sundquist WI, Kay MS, Shen PS, Hill CP. Structure of Vps4 with circular peptides and implications for translocation of two polypeptide chains by AAA+ ATPases (2019). Elife e44071.
-
Disotuar MM, Petersen ME, Nogueira JM, Kay MS, Chou DH. Synthesis of hydrophobic insulin-based peptides using a helping hand strategy (2019). Org Biomol Chem. 17:1703-1708.
-
Redman JS, Francis JN, Marquardt R, Papac D, Mueller AL, Eckert DM, Welch BD, Kay MS. Pharmacokinetic and Chemical Synthesis Optimization of a Potent D-Peptide HIV Entry Inhibitor Suitable for Extended-Release Delivery (2018). Mol Pharm. 15(3):1169-1179.
-
Jacobsen MT, Erickson PW, Kay MS. Aligator: A computational tool for optimizing total chemical synthesis of large proteins (2017). Bioorg Med Chem. 25(18):4946-4952.
-
Jacobsen MJ, Petersen ME, Ye X, Galibert M, Lorimer GH, Aucagne V, and Kay MS. A Helping Hand to Overcome Solubility Challenges in Chemical Protein Synthesis (2016). J Am Chem Soc. 138(36):11775-82.
-
Petersen ME, Jacobsen MT, Kay MS. Synthesis of tumor necrosis factor α for use as a mirror-image phage display target (2016). Org Biomol Chem. 14(23):5298-303.
-
Clinton TR, Weinstock MT, Jacobsen MT, Szabo-Fresnais N, Pandya MJ, Whitby FG, Herbert AS, Prugar LI, McKinnon R, Hill CP, Welch BD, Dye JM, Eckert DM, Kay MS. Design and characterization of ebolavirus GP prehairpin intermediate mimics as drug targets (2015). Protein Sci. 24:446-63.
-
Weinstock MT, Jacobsen MT, and Kay MS. Synthesis and folding of a mirror-image enzyme reveals ambidextrous chaperone activity (2014). PNAS 111: 11679–11684.
-
Mesquita PM, Srinivasan P, Johnson TJ, Rastogi R, Evans-Strickfaden T, Kay MS, Buckheit KW, Buckheit RW Jr, Smith JM, Kiser PF, Herold BC. Novel preclinical models of topical PrEP pharmacodynamics provide rationale for combination of drugs with complementary properties (2013). Retrovirology 10:113.
-
Pang HB, Hevroni L, Kol N, Eckert DM, Tsvitov M, Kay MS*, Rousso I*. Virion Stiffness Regulates Immature HIV-1 Entry (2013). Retrovirology 10:4.
-
Francis, JN, Redman, JS, Eckert, DM, and Kay MS. Design of a Modular Tetrameric Scaffold for the Synthesis of Membrane-Localized D-peptide Inhibitors of HIV-1 Entry (2012). Bioconjugate Chemistry 23: 1252-8.
-
Weinstock, MT, Francis, JN, Redman, JS, and Kay, MS (2012). Protease-Resistant Peptide Design – Empowering Nature’s Fragile Warriors Against HIV. Peptide Science, 98: 431-42.