Erik Blackwood
Assistant Professor of Nutrition & Integrative Physiology
Heart Failure, Cardiometabolic Disease, Atrial Myopathy, Cardiac Fibrosis, Inter-Organ Communication, Cardiokine Secretion, Cellular Proteostasis, ER Stress, Cardiac Metabolism, Gene Therapy

Molecular Biology Program
Education
B.S.The University of Notre Dame
Ph.D.San Diego State University
Research
Heart disease is the leading cause of mortality in the US and is often preceded by pathological cardiac hypertrophy due to chronic hypertension and sustained increases in cardiac afterload. While initially an adaptive response to maintain cardiac output and systemic blood supply, pathological cardiac hypertrophy eventually leads to a decompensated state of heart failure (HF). The initial adaptive aspect of hypertrophic growth requires increases in protein synthesis, which must be balanced by adequate protein folding, and degradation of misfolded, potentially toxic proteins. Without this balance, cardiac myocytes cannot maintain protein homeostasis, or proteostasis, which threatens functional cellular integrity and viability. The Blackwood laboratory is focused on elucidating novel mechanisms whereby proteostasis machinery in the heart protects against chronic systemic diseases, such as HF and global metabolic syndrome, translating these discoveries into novel therapeutics, and evaluating their efficacy in preclinical models of HF. Specifically, we have three directives guiding our research program: (1) Elucidate novel mechanisms for heart-directed inter-organ communication during metabolic syndrome (i.e. Axes of Cardiometabolic Health); (2) Determine mechanisms contributing to atrial dysfunction and remodeling during HF; and (3) Determine the contributions of resident cardiac fibroblasts to the pathogenesis of HF. Below, we highlight two major ongoing funded projects in the lab:
Project #1: Roles for SGK1 in Cardiometabolic Heart Failure with Preserved Ejection Fraction
Heart failure with preserved ejection fraction (HFpEF) has emerged as the greatest unmet medical need in cardiovascular medicine, comprising 50% of all HF cases, with a US prevalence of Ñ3 million. HFpEF is associated with high morbidity and mortality, with a 5-year survival after hospitalization worse than most cancers of ~35% and, unlike the better understood heart failure with reduced ejected fraction (HFrEF), there are no evidence-based therapies for HFpEF. HFpEF represents an entirely new disease threat because of the systemic metabolic and endocrine nature of its pathogenesis. And, while HFpEF is heterogeneous in its clinical presentation, arguably the most prevalent form is cardiometabolic HFpEF (cMet HFpEF), where the main comorbidities are obesity, type 2 diabetes, and hypertension. These same comorbidities are the major drivers of concomitant obesity-related diseases that includes nonalcoholic fatty liver disease (NAFLD), which is prevalent in more than 50% of HFpEF patients and has been demonstrated to be an independent predictor of all-cause mortality in HFpEF. This is of unique concern as the obesity epidemic continues to grow with current estimations that 45% of the world’s population is either overweight or obese. Our preliminary data support the notion that the heart is a primary regulator of global metabolic syndrome through the serine-threonine kinase, serum glucocorticoid kinase 1 (SGK1).
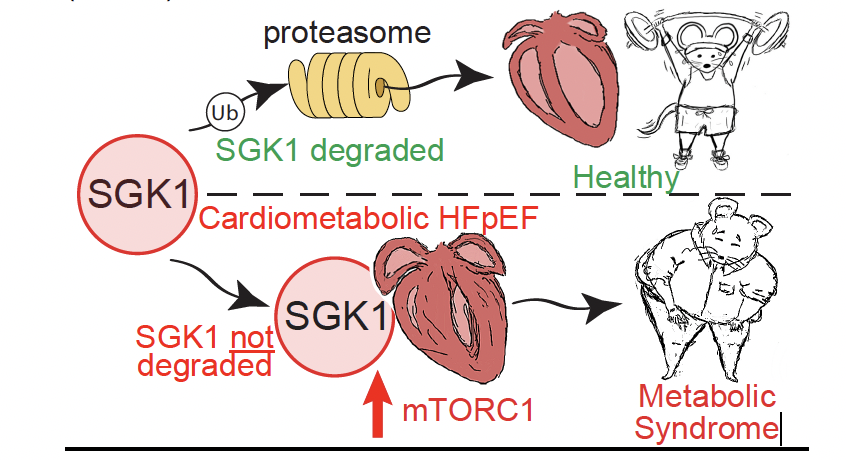
In this proposal, we will focus on cardiac-specific SGK1 in cMet HFpEF with the hypothesis that SGK1 promotes increased mTORC1 activity in the heart via phosphorylating the negative regulator of mTORC1, PRAS40, which exacerbates cardiac pathology and subsequent global metabolic dysfunction in cMet HFpEF. We will address this hypothesis using cardiac-specific targeting of SGK1 and PRAS40 in a mouse model of cMet HFpEF with a focus on assessments of cardiac function and metabolism, as well as mechanistic studies in primary cardiomyocytes, in the following Specific Aims which are to: (Aim 1) determine the role for cardiac SGK1-PRAS40 signaling in regulating heart function, metabolism, and heart directed inter-organ communication during cMet HFpEF, (Aim 2) determine whether PRAS40 inhibition by SGK1 promotes mTORC1 mediated pathologic cardiac metabolic inflexibility during cMet HFpEF, and (Aim 3) evaluate the therapeutic efficacy of a novel SGK1 inhibitory peptide for preserving cardiac function and systemic metabolism during cMet HFpEF. These studies are significant as the present the opportunity to identify novel secreted factors from the heart that regulate global metabolic health as well as to test new therapeutic targets for both HFpEF and obesity-related diseases.
Project #2: Novel Roles for IRE1 in Regulating Cardiac Fibrosis
Cardiovascular disease has consistently been the leading cause of death in the US for the last century with resultant heart failure (HF) accounting for nearly 800,000 deaths per year. Despite substantial advancements in clinically available interventions, the incidence of HF continues to rise with a staggering estimated economic burden of >$900 billion per year by 2030. Virtually all etiologies of cardiovascular disease leading to both heart failure with reduced ejection fraction (HFrEF) and preserved ejection fraction (HFpEF) involve pathological remodeling of the left ventricle characterized by excessive deposition of extracellular matrix (ECM) proteins that impair ventricular compliance and perpetuate HF pathogenesis. The primary cell type responsible for synthesizing and secreting ECM proteins are resident cardiac fibroblasts (CFBs) that become activated by diverse stimuli during HF. As a testament to the secretory potential of CFBs, a single fibroblast can produce >500,000 procollagen chains per hour which necessitates the presence of a robust network of protein folding machinery in the endoplasmic reticulum to maintain cellular proteostasis and allow for the proper processing of nascent ECM proteins. Such protein folding demands trigger activation of the unfolded protein response (UPR), an adaptive component of the proteostasis network to facilitate proper protein quality control. Our preliminary data support the notion that the IRE1 arm of the UPR plays a pivotal role in protecting against fibrotic remodeling in the heart via targeted mRNA degradation of transcripts encoding proteins required for CFB activation, namely Tmem100.
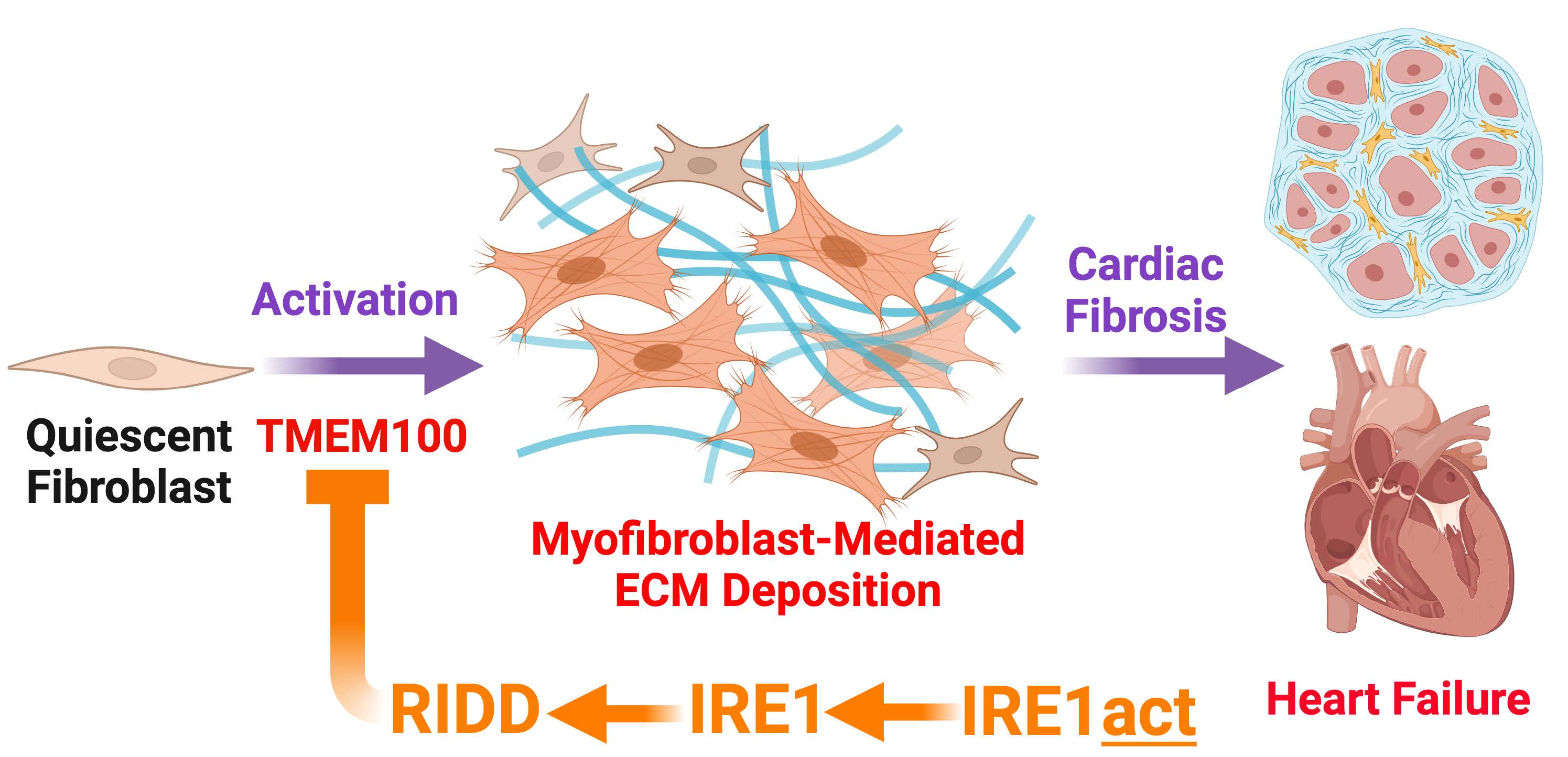
In this proposal, we will focus on IRE1 and whether tactile control of the endonuclease activity of IRE1 could alter CFB activation and ECM deposition with the hypothesis that IRE1 protects against pathological cardiac fibrosis in HF via regulating the selective degradation of Tmem100 and a pro-fibrotic transcriptome. We will address this hypothesis using fibroblast-specific gene targeting in complimentary mouse models of HF, as well as mechanistic studies in primary CFBs, in the following Specific Aims which are to: (Aim 1) determine the cardiac fibroblast transcriptomic profile regulated by IRE1 using a mouse model of HFrEF, (Aim 2) determine the functional significance of IRE1-mediated degradation of Tmem100 mRNA in fibrotic remodeling using a mouse model of HFrEF, and (Aim 3) evaluate the therapeutic efficacy of novel small molecule activator of IRE1 in mitigating fibrotic remodeling using mouse models of HFrEF or HFpEF. These studies are significant as they present the opportunity to identify novel mechanisms contributing to CFB activation and ECM deposition as well as to test new therapeutic strategies with potential to ameliorate fibrotic remodeling associated with both HFrEF and HFpEF.
References (Selected Publications)
- Hale, T. M., & Blackwood, E. A. (2024). Modeling heart failure with preserved ejection fraction in female mice: an elusive target. American journal of physiology. Heart and circulatory physiology, 326(6), H1402–H1405.
- Hofmann, C., Aghajani, M., Alcock, C. D., Blackwood, E. A., Sandmann, C., Herzog, N., Groß, J., Plate, L., Wiseman, R. L., Kaufman, R. J., Katus, H. A., Jakobi, T., Völkers, M., Glembotski, C. C., & Doroudgar, S. (2024). ATF6 protects against protein misfolding during cardiac hypertrophy. Journal of molecular and cellular cardiology, 189, 12–24.
- Bilal, A. S., Parker, S. N., Murray, V. B., MacDonnell, L. F., Thuerauf, D. J., Glembotski, C. C., & Blackwood, E. A. (2023). Optimization of Large-Scale Adeno-Associated Virus (AAV) Production. Current protocols, 3(5), e757.
- Blackwood, E. A., MacDonnell, L. F., Thuerauf, D. J., Bilal, A. S., Murray, V. B., Bedi, K. C., Jr, Margulies, K. B., & Glembotski, C. C. (2023). Noncanonical Form of ERAD Regulates Cardiac Hypertrophy. Circulation, 147(1), 66–82.
- Bilal, A. S., Blackwood, E. A., Thuerauf, D. J., & Glembotski, C. C. (2021). Optimizing Adeno-Associated Virus Serotype 9 for Studies of Cardiac Chamber-Specific Gene Regulation. Circulation, 143(20), 2025–2027.
- Blackwood, E. A., Bilal, A. S., Azizi, K., Sarakki, A., & Glembotski, C. C. (2020). Simultaneous Isolation and Culture of Atrial Myocytes, Ventricular Myocytes, and Non-Myocytes from an Adult Mouse Heart. Journal of visualized experiments : JoVE, (160), 10.3791/61224.
- Blackwood, E. A., Thuerauf, D. J., Stastna, M., Stephens, H., Sand, Z., Pentoney, A., Azizi, K., Jakobi, T., Van Eyk, J. E., Katus, H. A., Glembotski, C. C., & Doroudgar, S. (2020). Proteomic analysis of the cardiac myocyte secretome reveals extracellular protective functions for the ER stress response. Journal of molecular and cellular cardiology, 143, 132–144.
- Blackwood, E. A., Azizi, K., Thuerauf, D. J., Paxman, R. J., Plate, L., Kelly, J. W., Wiseman, R. L., & Glembotski, C. C. (2019). Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nature communications, 10(1), 187.
- Blackwood, E. A., Hofmann, C., Santo Domingo, M., Bilal, A. S., Sarakki, A., Stauffer, W., Arrieta, A., Thuerauf, D. J., Kolkhorst, F. W., Müller, O. J., Jakobi, T., Dieterich, C., Katus, H. A., Doroudgar, S., & Glembotski, C. C. (2019). ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circulation research, 124(1), 79–93.
- Jin, J. K., Blackwood, E. A., Azizi, K., Thuerauf, D. J., Fahem, A. G., Hofmann, C., Kaufman, R. J., Doroudgar, S., & Glembotski, C. C. (2017). ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circulation research, 120(5), 862–875.